LEMMA
Example data
The example directory contains a simulated dataset with:
- 5000 individuals
- 20,000 SNPs
- 5 environmental variables
- a simulated phenotype
The phenotype has been simulated to have:
- 2000 SNPs with non-zero main effects
- 500 SNPs with non-zero GxE effects
- GxE effects with a linear combination of two of the five environments (i.e. two are active).
- SNP-Heritability of 20% (main effects) and 10% (GxE effects)
Getting started
The LEMMA approach consists of three distinct steps:
- A variational inference algorithm computes the Environmental Score (ES) and residualised phenotype. This is typically run on genotyped SNPs.
- Single SNP association testing using the ES and residualised phenotypes. This can be run either on the same set of genotyped SNPs, or a larger set of imputed SNPs.
- Heritability estimation partitioned into additive SNP effects and multiplicative GxE effects with the ES.
All three steps can be run in sequence using the following commands
rm example/bgen_filenames.txt
for cc in `seq 1 22`; do
echo "example/n5k_p20k_example_chr${cc}.bgen" >> example/bgen_filenames.txt;
done
mpirun build/lemma_1_0_4 \
--pheno example/pheno.txt.gz \
--environment example/env.txt.gz \
--VB \
--bgen example/n5k_p20k_example.bgen \
--singleSnpStats \
--RHEreg --random-seed 1 \
--mStreamBgen example/bgen_filenames.txt \
--out example/inference.out
For association testing and heritability estimation, LEMMA will use genetic data provided from the --mStreamBgen if it is provided. Otherwise LEMMA will use genetic data from the --bgen flag.
Files provided to --mStreamBgen should each contain only one chromosome. Separating the chromosomes into different files can be achieved with the BGENIX program.
Output from the variational inference algorithm:
example/inference.out.gz: converged hyperparameter values + ELBOexample/inference_converged_eta.out.gz: converged Environmental Scoreexample/inference_converged_resid_pheno_chr*.out.gz: residual phenotype per chromosomeexample/inference_converged_vparams_*.out.gz: posterior variational parametersexample/inference_converged_yhat.out.gz: predicted vectors and residualised phenotypes
Output from association testing:
example/inference_loco_pvals.out.gz
Output from heritability estimation:
example/inference_pve.out.gz
The LEMMA algorithm is modular, and so each step can be performed separately as follows.
Running the LEMMA variational inference algorithm
mpirun build/lemma_1_0_4 \
--VB \
--pheno example/pheno.txt.gz \
--environment example/env.txt.gz \
--bgen example/n5k_p20k_example.bgen \
--out example/inference.out
In this case the algorithm should converge in 59 iterations.
Association testing with imputed SNPs
mpirun build/lemma_1_0_4 \
--singleSnpStats --maf 0.01 \
--pheno example/pheno.txt.gz \
--resid-pheno example/inference_converged_yhat.out \
--mStreamBgen example/bgen_filenames.txt \
--environment example/inference_converged_eta.out \
--out example/inference_loco_pvals.out
In this example the flag --pheno example/pheno.txt.gz is optional. This is used to see if any environmental variables have significant squared effects, and include them as covariates if so.
For analyses of large genomic datasets it may be useful to parallelize association testing across chunks of SNPs with the --range flag.
Heritability estimation
mpirun build/lemma_1_0_4 \
--RHEreg --random-seed 1 \
--pheno example/pheno.txt.gz \
--mStreamBgen example/bgen_filenames.txt \
--environment example/inference_converged_eta.out \
--out example/inference_pve.out
This should return heritability estimates of h2-G = 0.23 (0.032) and h2-GxE = 0.08 (0.016), where the value in brackets is the standard error.
Advanced usage
Parallelism with OpenMPI
LEMMA performs parallel processing with OpenMPI, and does so using the SPMD (Single Process Multiple Data) paradigm.
More explicitly, samples are partitioned such that blocks of rows of the phenotype y,
genotypes X and environmental variables E are assigned to each core. Each core then runs inference only on the locally held block of samples. At relevant points in the algorithm, cores then message summary-level statistics to each other, such that the algorithm is invariant to the number of cores; or rather, we would get the same result by loading all of the data onto only one core.
When running LEMMA on the UK Biobank we found parallelising with OpenMPI to highly efficient (ie doubling the number of cores almost doubles computational speed) up to when the number of samples per core is a couple of thousand, after which adding extra cores yielded diminishing returns. Using OpenMPI has the additional advantage of allowing users to utilise cores from across a cluster rather than being restricted to a single node.
To set the number of cores on the commandline explicitly, use
mpirun -n <cores> build/lemma_1_0_4
Precomputing the dXtEEX array
Before running the variational algorithm, LEMMA requires the quantities
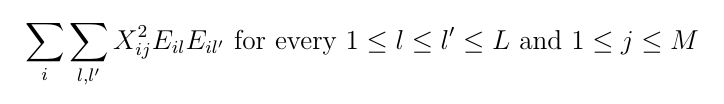
LEMMA is able to compute this internally, however for large datasets this imposes substantial costs. As this is easily parallelised over variants and/or environments, we recommend that users precompute this quantity beforehand and provide a file to LEMMA at runtime.
Install bgen_utils using instructions from https://github.com/mkerin/bgen_utils.
Build the example/dxteex.out.gz array using commands
BGEN_UTILS=<path_to_bgen_utils>
for cc in `seq 1 22`; do
${BGEN_UTILS} \
--compute-env-snp-correlations \
--mode_low_mem \
--range ${cc}:0-100000000000 \
--bgen $(bgen) \
--environment $(dir)/env.txt \
--out example/dxteex_chr${cc}.out.gz;
done
zcat example/dxteex_chr*.out.gz > example/dxteex.out.gz
Then provide the file example/dxteex.out.gz to LEMMA with the commandline flag --dxteex.
mpirun build/lemma_1_0_4 \
--VB \
--pheno example/pheno.txt.gz \
--environment example/env.txt.gz \
--bgen example/n5k_p20k_example.bgen \
--dxteex example/dxteex.out.gz \
--out example/inference.out
Heritability partitioned by MAF and LD
For this you will need:
- LD-scores (we recommend using GCTA with a window of size
--ld-wind 1000) - MAF of each SNP obtained from the UKBiobank MFI files.
To convert into the file format expected by LEMMA we have provided a brief Rscript scripts/preprocess_ldms_groups.R.
Then run the heritability analysis as follows
mpirun build/lemma_1_0_4 \
--RHEreg --n-RHEreg-samples 20 --n-RHEreg-jacknife 100 --random-seed 1 \
--pheno example/pheno.txt.gz \
--bgen example/n5k_p20k_example.bgen \
--environment example/inference_converged_eta.out \
--RHEreg-groups example/ldms_groups.txt \
--out example/rhe_ldms.out
Resuming from a previous parameter state
In case of runtime crashes, LEMMA can save the parameter state at periodic intervals by providing the commandline flag --resume-from-state. LEMMA can then subsequently resume inference from this saved state. For example
mpirun build/lemma_1_0_4 \
--VB \
--pheno example/pheno.txt.gz \
--environment example/env.txt.gz \
--bgen example/n5k_p20k_example.bgen \
--state-dump-interval 10 \
--out example/inference.out
mpirun build/lemma_1_0_4 \
--VB \
--pheno example/pheno.txt.gz \
--environment example/env.txt.gz \
--bgen example/n5k_p20k_example.bgen \
--resume-from-state example/lemma_interim_files/inference_dump_it30 \
--out example/inference_from_it30.out
diff example/inference_from_it30.out example/inference.out
Outputs from the two should match, up to some small numerical difference in the ELBO. Note that if the iteration number that you start from is not a multiple of 3, then output will not match exactly because the SQUAREM algorithm adapts the trajectory of the hyperparameter updates in multiples of three.
Complexity
Computational Complexity
LEMMA uses a iterative algorithm to approximate the posterior distribution. The per-iteration complexity is O(NM).
Memory Complexity
To store the genotype matrix, LEMMA uses approximately MN bytes of RAM where M is the number of genotyped SNPs and N is the number of samples.
To store an array of SNP-environment correlations, LEMMA uses a further 8ML(L+1)/2 bytes of RAM, where L is the number of environments and M is the number of SNPs. For M = 600k and L < 100 this should not be a dominating requirement.